Genetics and genomics for wheat improvement
Our lab aims to understand gene function in wheat, with a view to improving the nutritional value of the grain. This goal is complicated by the polyploid nature of the wheat genome so we also work on the effects of polyploidy on gene expression and phenotype in wheat.
Why does polyploidy matter?
Wheat is a polyploid species, therefore it has multiple copies of each gene, known as homoeologs. In general, we do not know if these copies have the same or different biological roles. If the homoeologs have the same biological role they are said to be functionally redundant and all the homoeologs would need to be targeted for crop improvement. Therefore our lab aims to understand how homoeologs influence wheat biology and to use this knowledge to improve the nutrient content of wheat. To do this we use a wide range of techniques: from genetics, genomics and bioinformatics to greenhouse experiments and molecular biology.
For a general background to our work check out this presentation aimed at undergraduate students explaining why polyploids like wheat matter:
Areas of interest:
1) Genes regulating wheat nutrient content
Wheat provides over 20 % of calories eaten by humankind globally, but it is also an important source of protein and micronutrients in the human diet. In fact wheat provides over 20 % of protein eaten by mankind which is more than the contribution made by eating meat.
We need to increase crop yields to feed the world’s growing population, but unfortunately increasing yields usually results in a decrease in nutrient content which could have adverse health outcomes.
We are using molecular biology, plant physiology, genomics and computational approaches to understand the genes regulating senescence (ageing) and nutrient remobilisation with the aim to maintain or increase nutrient content alongside increasing yields.
Transcription factors regulating senescence
Although senescence strongly affects the final protein and micronutrient content of wheat grain, little is known about the genetic regulation of this coupled process of senescence and nutrient remobilisation. We have used gene regulatory network modelling (Borrill et al., 2019) and time-course transcriptomics (Andleeb et al., 2022) to identify novel transcription factors which are candidates to regulate senescence in wheat. We are currently validating their function using TILLING mutants and starting to characterise their downstream targets.
To find out more about how we identified transcription factors regulating senescence (and why gene networks are a powerful tool in polyploids like wheat) please take a look at this webinar:
Iron and zinc content

Malnutrition affects millions of people worldwide. This suffering could be alleviated by the production of crops with higher levels of micronutrients such as iron and zinc. Recently many new genomic resources have been made available for wheat that could be used for biofortification (reviewed in Ali and Borrill, 2020). Using these resources, we aim to identify genes to use in wheat biofortification. These genes could help breeders produce wheat varieties with high levels of iron and zinc in their grain. Currently we are assessing whether induced variation can be used to create wheat lines with higher iron and zinc levels.
The use of gene editing is accelerating progress in this important topic, as Philippa recently discussed with Brian Cox:
2) Wheat as a model to understand polyploid genome regulation
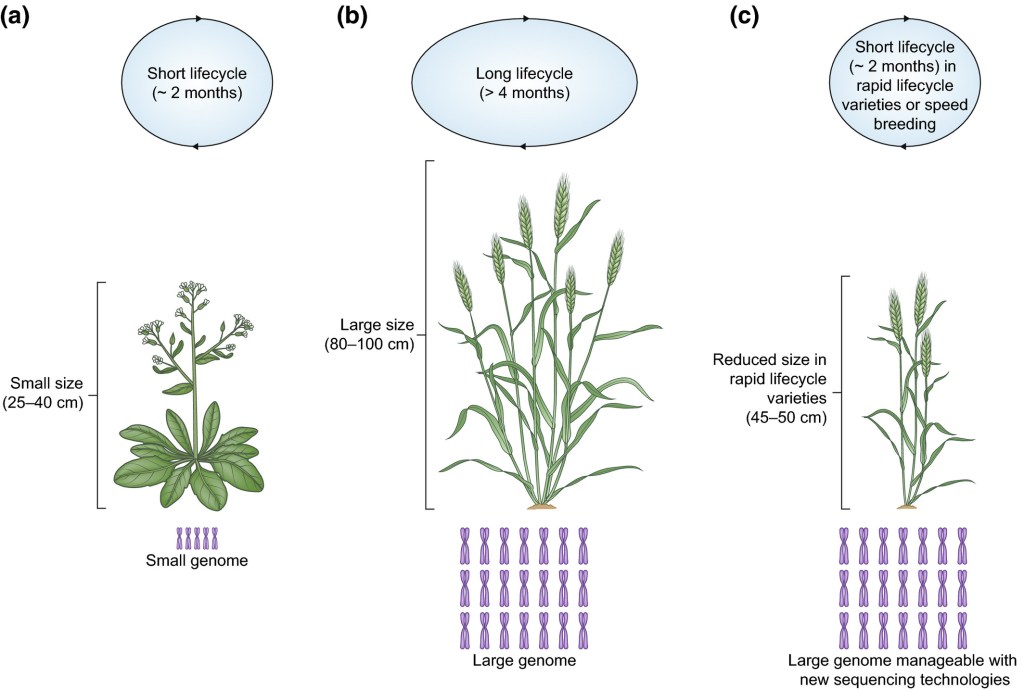
The most widely grown species of wheat (Triticum aestivum) is an allohexaploid, with three homoeologous copies of most genes, which together constitute a triad. Within each triad, the three homoeologs are very similar (>95 % identical within coding sequences). Changes in one homoeolog (e.g. a knock-out mutation) may be hidden by functional redundancy with the other homoeologs (reviewed in Borrill, Harrington and Uauy, 2019). If we could understand and overcome this functional redundancy, we could more easily alter agronomic traits in polyploid crops such as wheat to address the challenge of food security. However, currently we have limited understanding of how many genes in the wheat genome have functionally redundant homoeologs, the mechanisms influencing functional redundancy and how to avoid functional redundancy for trait improvement.
During the last 5 years, through a series of international collaborations, we have developed key resources for wheat functional genomics including a gene expression atlas (Borrill, Ramírez-González and Uauy., 2016; Ramírez-González, Borrill et al., 2018; www.wheat-expression.com), a fully annotated genome assembly (IWGSC, 2018) and sequenced mutant populations to characterise gene function (Krasileva et al., 2017). Together these resources lay the foundation to use wheat as an experimentally tractable polyploid model species (Borrill, 2020).
Mechanisms controlling homoeolog expression in polyploid wheat
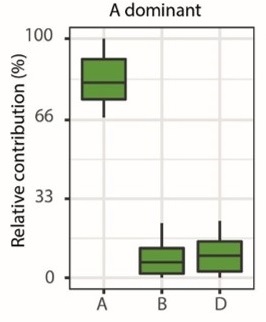
Several major agronomic traits are controlled by changes in the expression level of one homoeolog including VRN1 and PPD1 that have been essential to adapt wheat flowering to a range of environmental conditions. These well-studied genes are not unique in showing differences in the expression levels of homoeologs. We recently found that homoeologs in ~30% of triads differ in their expression levels, i.e. one homoeolog is over or under expressed compared to the other two gene copies (Ramírez-González, Borrill et al., 2018).
You can watch a presentation about our research in this area here:
We have recently started a project funded by BBSRC to explore the genetic and epigenetic factors which influence homoeolog expression.We hope to discover fundamental information about the control of gene expression in polyploid wheat and identify potential routes to manipulate homoeolog expression that may also be applicable to other polyploid crops of agronomic importance.
Resources
We also generate wheat genomics resources including gene expression atlases, sequenced mutant populations and “how to guides” to explain resources that are available. Find out more here.
Code
We believe in making our code open so that others can re-produce our analyses and build upon our work. Find our code on our Github page.
Funding
We are grateful to the funders who have supported our research:

